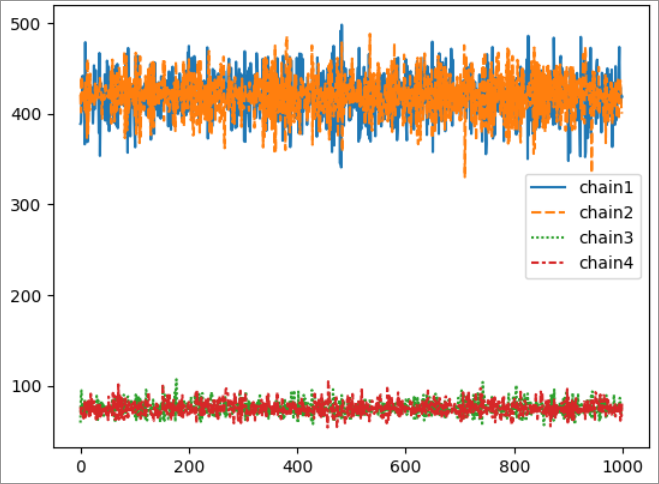
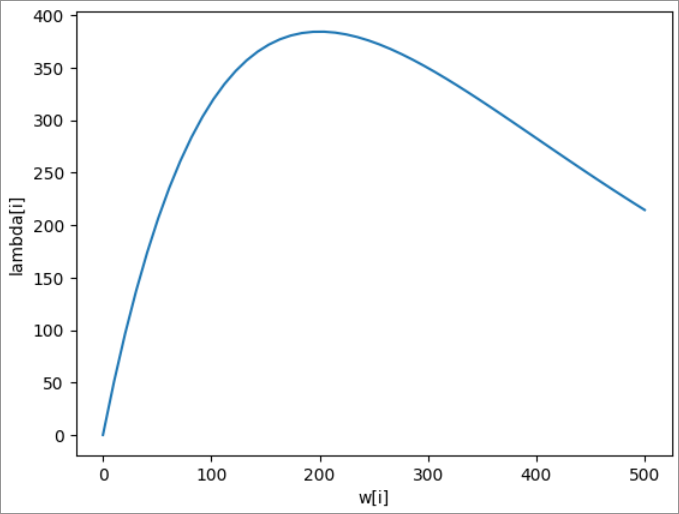
I am brand new to Bayesian inference, Stan and Rstan. I am attempting to infer individual-level worm burdens based on individual-level egg outputs, initially just with very simple dummy data. I have generated dummy data:
#generate dummy data for worm counts, based on a negative binomial distribution
worms <- rnbinom(1000, size = 0.5, mu = 20)
#density dependence of fecundity in worms
lambda <- ifelse(worms > 0, 5.2*worms*exp((worms - 1)*-0.005), 0)
#now use worm data to generate egg counts per individual
eggs <- rpois(length(worms), lambda)
I then set up a simple stan model:
// data block
data {
int<lower = 0> N;
array[N] int<lower = 0> egg_count; // egg count data
real<lower = 0> fec; // worm fecundity
real dd; // density dependence term
}
// parameter block
parameters {
array[N] real<lower = 0> worms; // estimate worm number
}
// model block
model {
// no priors
// likelihood function
for (i in 1:N) {
egg_count[i] ~ poisson(fec*worms[i]*exp((worms[i] - 1)*dd)); // egg count is poisson distributed based on worms * fecundity * density dependence
}
}
I then run the model using RStan:
#prepare dataset to pass to stan
worm_data <- list(N = length(eggs),
egg_count = eggs,
fec = 5.2,
dd = -0.005)
#run model
wb.model <- stan(
file = "X.stan",
data = worm_data,
iter = 30000,
chains = 3,
verbose = FALSE
)
Weirdly, the model is able to infer worm burdens well at low egg count values, but does not converge at higher egg count values. I have tried increasing the iterations on the model, but it still does not converge.
What am I doing wrong?
Thanks in advance for the help.