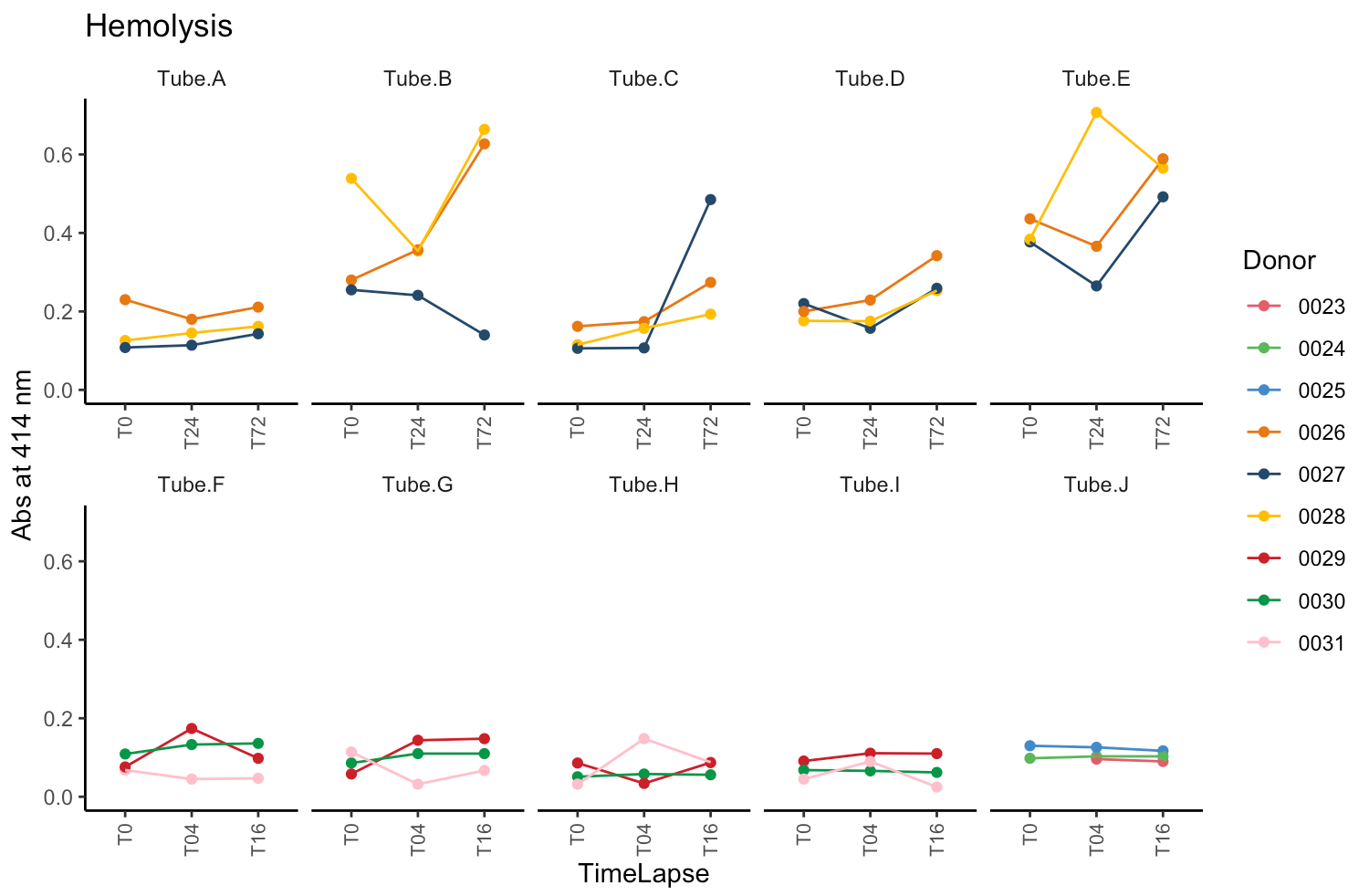
Hi Stan community,
I’m a grad student and got a dataset which I would like to model in brms. I’ve read Statistical Rethinking and been following Frank Harell his lessons, but I’m having difficulties to implement this to my own topics. I’m new to all of this and haven’t had formal statistical training, so please bear with me if I ask (too) obvious questions.
The dataset measured hemolysis in 8 different blood tubes (lets label them A-H), and we want to evaluate which tube has the lowest and highest hemolysis. For tube A-D, we drew blood from 3 volunteers, and for tube F-H, we drew blood in 3 other volunteers. Furthermore, for every tube we have a measurement at timepoint 0 (T0) and later timepoints (T04 for tube A-D, T16 for tube A-D, T24 for tube F-H and T72 for tube F-H).

The resulting dataset & code looks like this:
require(brms)
require(bayesplot)
tubes <- c("A","A","A","B","B","B")
donors <- c("Donor1","Donor1","Donor1", "Donor1", "Donor1", "Donor1")
hemolysis <- c(0.10,0.34,0.12,0.23,0.45, 0.56)
timepoint <- c("T0", "T04", "T16", "T0", "T04", "T16")
hemolysis_df <- as.data.frame(cbind(tubes, donors, hemolysis,timepoint))
# I tried to specify here a sceptical prior that all tubes suffer from hemolysis based on prior knowledge in literature (value of > 0.5 indicates hemolysis)
prior <- c(prior_string("normal(0.5,0.1)", class = "b"))
# Sampling from prior, I set the family to lognormal so the results are non-negative
fit <- brm(formula = hemolysis ~ tube + tube:timepoint + (1 | donor), data = hemolysis_df, family = lognormal(), chains = 4, cores = 4, iter = 2000, prior = prior, sample_prior = "only")
# Checking output
summary(fit)
plot(fit)
# Sampling
fit_model2 <- update(fit, sample_prior = "no", chains = 4, cores = 4, iter = 2000, control = list(adapt_delta = 0.90, max_treedepth = 13))
# Checking results
summary(fit_model2)
plot(fit_model2)
prior_summary(fit_model2)
marginal_effects(fit_model2)
pp_check(fit_model2)
I found this post ((More) Advice on a Multilevel Time Series Model in BRMS) which looks to have a similar set-up, but if I try to model it like that I"m getting an error “term has fewer unique covariate combinations than specified maximum degrees of freedom”, probably because the dataset is too small.
My questions:
- Is this dataset even suitable to do statistical modelling (because there are only 3 observations/donor per timepoint + tube combination?
- How can I incorporate the donor and time effect in the model?
- Should the timepoints be coded categorical (as factors) or continuous (as numeric)?
- Is my code decent? Is the prior correctly defined?
- Is this the right place to ask for help like this, or are there better places?