I am fitting a nonlinear mixed-effects model to longitudinal data.
The response variable I am trying to model is the hydrodynamic radius of a an aggregating lipoprotein. The values for the radius are obtained through dynamic light scattering (DLS).
The reason I am explaining this is because the instrument used to measure this radius has a “limit”, after which the measurements are not guaranteed to be very accurate.
The values of the radius measured vary between 15-5000nm.
The recommendation is to usually use values up to a 1000, or slightly more.
So I decided to try out and fit my model twice - once with the whole dataset (with values up to 5000), let’s call it Model1, and once only with values up to 1500 - Model2.
*Note that Model1 and Model 2 are essentially the same model (same fixed and random effects), but priors adjusted for the values in the dataset.
The entire code for the model is given at the end of this post.
The result I got was interesting.
Model1
Family: gaussian
Links: mu = identity; sigma = identity
Formula: LDL ~ 15 + (alpha - 15)/(1 + exp((gamma - Time)/delta))
alpha ~ 1 + (1 | Cat) + (1 | Sample)
gamma ~ 1 + (1 | Cat) + (1 | Sample)
delta ~ 1
Data: Data (Number of observations: 527)
Samples: 3 chains, each with iter = 3000; warmup = 1000; thin = 1;
total post-warmup samples = 6000Group-Level Effects:
~Cat (Number of levels: 2)
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sd(alpha_Intercept) 16.22 12.26 0.64 45.96 5969 1.00
sd(gamma_Intercept) 0.44 0.18 0.09 0.81 1768 1.00~Sample (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sd(alpha_Intercept) 23.09 17.31 0.89 65.13 3073 1.00
sd(gamma_Intercept) 0.48 0.04 0.42 0.56 930 1.00Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
alpha_Intercept 4229.66 122.64 4006.64 4478.77 3687 1.00
gamma_Intercept 2.88 0.19 2.51 3.22 1702 1.00
delta_Intercept 0.44 0.02 0.41 0.47 3487 1.00Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sigma 382.00 12.29 358.80 407.40 5401 1.00
As one can see, sigma is quite big, which means there would be a lot of uncertainty for the predicted values.
As to the loo output
Computed from 6000 by 527 log-likelihood matrix
Estimate SEelpd_loo -3943.9 47.1
p_loo 99.6 12.1
looic 7887.9 94.2
Monte Carlo SE of elpd_loo is NA.
Pareto k diagnostic values:
Count Pct. Min. n_eff
(-Inf, 0.5] (good) 483 91.7% 833
(0.5, 0.7] (ok) 32 6.1% 220
(0.7, 1] (bad) 10 1.9% 22
(1, Inf) (very bad) 2 0.4% 4
See help(‘pareto-k-diagnostic’) for details.
Warning message:
Found 12 observations with a pareto_k > 0.7 in model ‘model’. With this many problematic observations, it may be more appropriate to use ‘kfold’ with argument ‘K = 10’ to perform 10-fold cross-validation rather than LOO.
Now, 12 bad observations is already a bit worrying, so I looked at the p_loo as well.
The number of parameters I get with parnames() is 133, which means
p_loo < Number of parameters, which, according to this means that my model is likely to be misspecified.
I also run a pp_check, which seemed to reflect the conclusions drawn from loo().

Then I looked at Model2, which was fit only using a subset of the dataset (with values considered to be more accurately measured by the instrument).
Here are the results I got:
Model2
Family: gaussian
Links: mu = identity; sigma = identity
Formula: LDL ~ 15 + (alpha - 15)/(1 + exp((gamma - Time)/delta))
alpha ~ 1 + (1 | Cat) + (1 | Sample)
gamma ~ 1 + (1 | Cat) + (1 | Sample)
delta ~ 1
Data: ldlData (Number of observations: 398)
Samples: 3 chains, each with iter = 6000; warmup = 1000; thin = 1;
total post-warmup samples = 15000Group-Level Effects:
~Cat (Number of levels: 2)
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sd(alpha_Intercept) 15.78 12.10 0.50 44.66 15151 1.00
sd(gamma_Intercept) 0.31 0.19 0.02 0.72 5022 1.00~Sample (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sd(alpha_Intercept) 183.34 16.29 152.78 216.58 3607 1.00
sd(gamma_Intercept) 0.44 0.04 0.38 0.52 2315 1.00Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
alpha_Intercept 1762.20 61.39 1644.00 1884.46 3464 1.00
gamma_Intercept 2.62 0.15 2.35 2.94 7811 1.00
delta_Intercept 0.23 0.01 0.22 0.24 5141 1.00Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Eff.Sample Rhat
sigma 39.04 2.29 34.56 43.53 3167 1.00
Note that this model was fit using longer chains, however, when using chains with equal length, I got very similar results.
As to the loo output:
Computed from 15000 by 398 log-likelihood matrix
Estimate SEelpd_loo -2161.9 36.6
p_loo 182.5 23.7
looic 4323.8 73.2Monte Carlo SE of elpd_loo is NA.
Pareto k diagnostic values:
Count Pct. Min. n_eff
(-Inf, 0.5] (good) 297 74.6% 1116
(0.5, 0.7] (ok) 20 5.0% 301
(0.7, 1] (bad) 55 13.8% 34
(1, Inf) (very bad) 26 6.5% 2
See help(‘pareto-k-diagnostic’) for details.
Warning message:
Found 81 observations with a pareto_k > 0.7 in model ‘model’. With this many problematic observations, it may be more appropriate to use ‘kfold’ with argument ‘K = 10’ to perform 10-fold cross-validation rather than LOO.
As one can see, the number of bad observations is really high.
In addition, the number of parameters I obtained with parnames() is 133,
so I have
Number of par. < p_loo, which according to this means that my model is badly misspecified.
I also run a pp_check, the plot of which is given below:
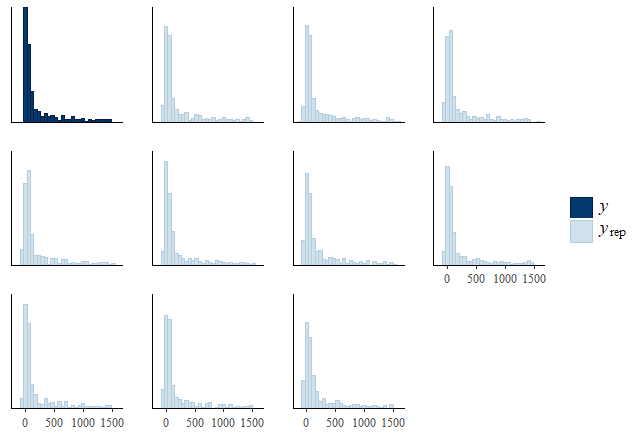
This looks pretty ok to me, much better than the pp_check plots of Model1.
However, in the above link it also said that if p_loo > N/5
(where N is the number of observations) then it is likely for pp_check to not detect the problem, which is probably true in my case.
So I have several questions:
1. Is Model2 badly misspecified?
2. If so, how come Model1, which is essentially the same (with the means and variances of my priors adjusted for the different datasets used), has less pareto_k > 0.7 datapoints?
3. Is it possible for my Model2 to still be useful, but just have higher number of influential observations, due to essentially “cutting out” the higher than 1500 values, essentially leaving the set of data points for each subject looking like this:

which potentially means that potentially the circled point influences the model strongly?
I forgot to mention that the predictions I obtain from Model2 are actually quite reasonable. In addition, I run the model with sample_prior=“only”, and I got no divergences, reasonable Eff. Sample, and Rhats=1.00
The code I’ve used for the model is given below.
fit_model ← function(Data){
fit<- brm(
bf(LDL ~ 15 + (alpha-15) / (1 + exp ((gamma-Time) / delta)),
alpha ~ 1 + (1|Cat) + (1|Sample) ,
gamma ~ 1 + (1|Cat) + (1|Sample),
delta ~ 1,
nl = TRUE),
prior = c(
prior(normal(1500, 30), class=“b”,nlpar = “alpha”),
prior(normal(2.5,0.07), class=“b”, nlpar = “gamma”),
prior(normal(0.2,0.05), class=“b”, lb=0, ub=0.5, nlpar = “delta”),
prior(student_t(3, 0, 10), class=“sigma”),
prior(student_t(3, 0, 10), class=“sd”, nlpar=“alpha”),
prior(student_t(3, 0, 10), class=“sd”, nlpar=“gamma”),
prior(normal(0.4,0.03 ), class=“sd”, group=“Sample”, coef=“Intercept”, nlpar=“gamma”),
prior(normal(300,10), class=“sd”, group=“Sample”, coef=“Intercept”, nlpar=“alpha”),
prior(normal(0.2,0.15), class=“sd”, group=“Cat”, coef=“Intercept”, nlpar=“gamma”),
prior(normal(17,6), class=“sd”, group=“Cat”, coef=“Intercept”, nlpar=“alpha”)),
data = Data, family = gaussian(),
chains = 3,
iter=3000,
warmup = 1000,
cores = getOption(“mc.cores”,1L),
thin = 1,
control = list(adapt_delta = 0.99999, stepsize=0.00001, max_treedepth=15)
)return(fit)
}
I am not sure if I managed to explain the problem clearly enough, but if anyone is willing to give me some advice, I will happy to answer further questions.