Almost always when I work with count data, NB seems to be better than Poisson. But recently I have an interesting problem, where it seems that the Poisson could, oddly, be better. I can’t share the data, but will share the posterior predictive checks, LOO, and grouped K-fold CV results.
I am analyzing an experiment where I have bacteria samples taken from different objects (object). Many experiments (experiment_id) are done for each of many object. For each experiment_id, two control samples are taken, two different treatments applied (experiment_type), and then two post-treatment samples are taken. Thus for each experiment_type there is a matched pair (control and treatment), pair_id. These are ‘within-object’ such that the object serves as it’s own control. The goal is to compare the two treatments.
The model formula is specified like this in brms:
bf(count ~ 1 + treatment + experiment_type + treatment:experiment_type + (1|object/experiment_id/pair_id))
I am interested in the interaction.
My question here is however about NB vs Poisson choice.
I fit one model, m1nb, using family=negbinomial, and one model, m1po, using family=poisson.
The results were very similar, though certainly not the same. Of note, the Poisson model required greater tree_depth and more iter, while the NB model ran fine with defaults on iter and tree_depth.
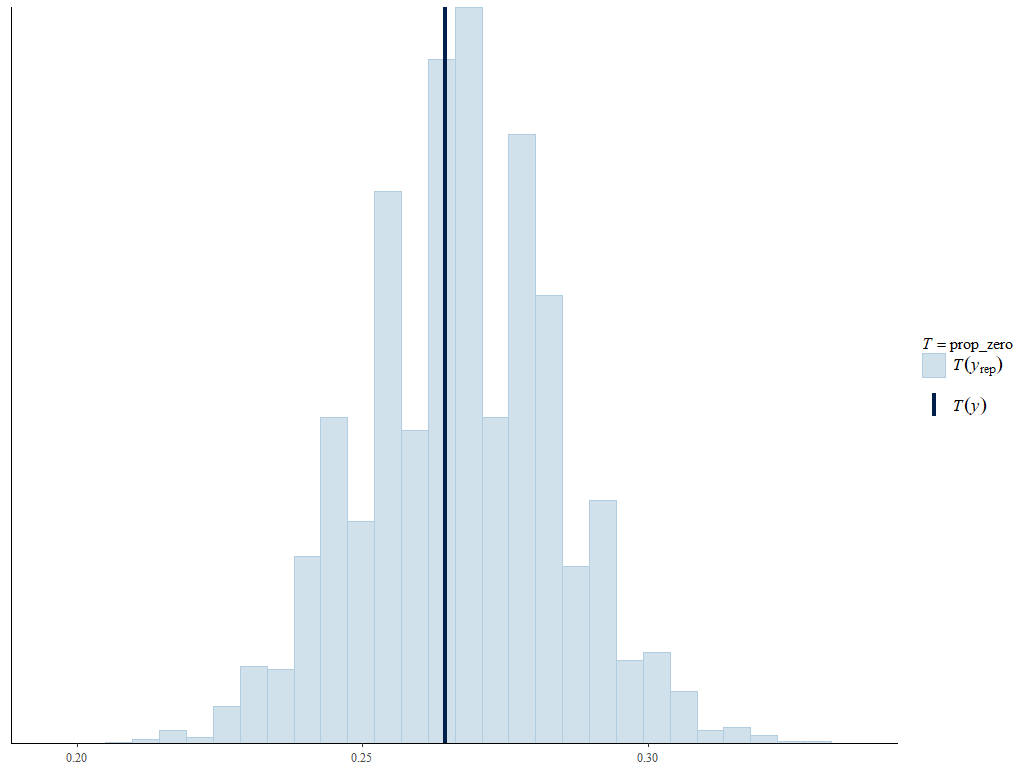
Viewing the posterior predictive checks, the Poisson model looks better.
Poisson:

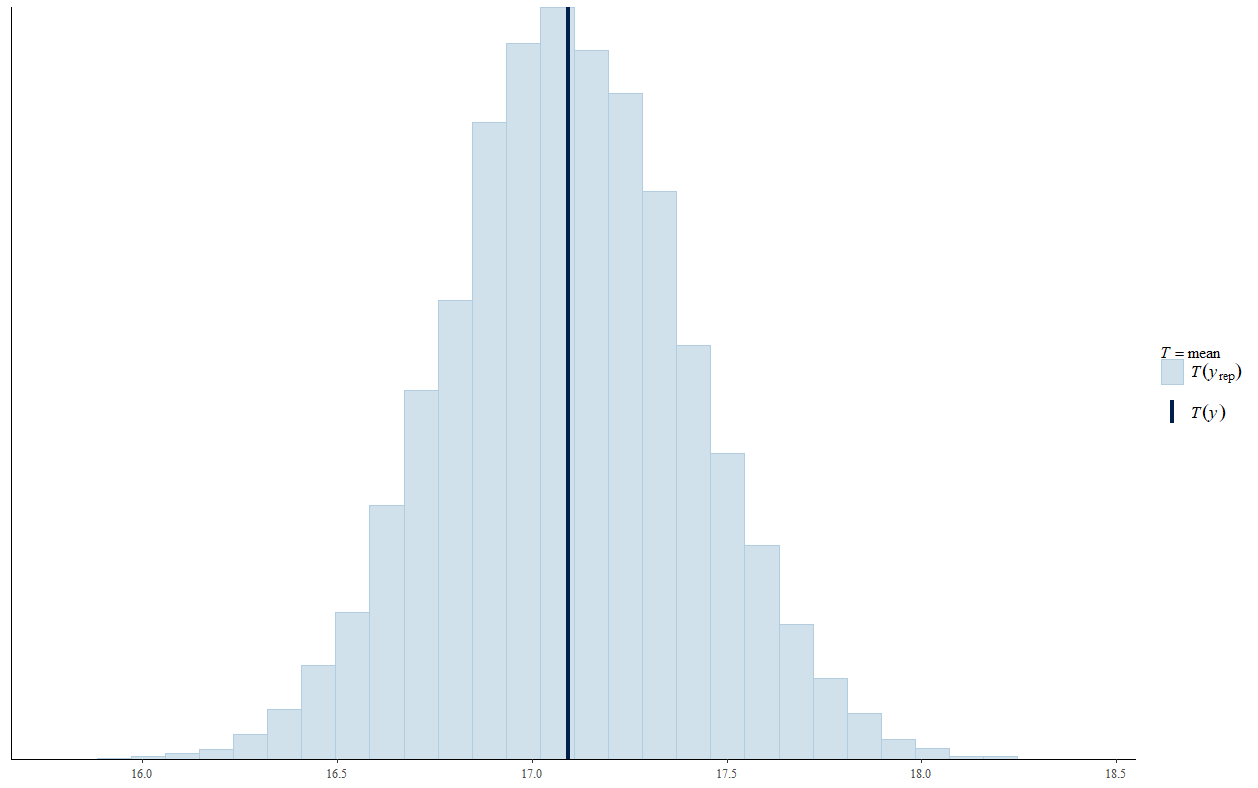
Negative binomial:


The negative binomial model posterior predictive checks show much higher max counts than seen in the data, and the mean is a little off.
I then ran LOO-CV and obtained these results:

There are many high pareto k’s, especially with the Poisson model. The negative binomial is favored. Referencing the helpful LOO FAQ, I can see that my case here is like the third example in part 16 of the FAQ, where for the Poisson model, p_loo > p and p > N/5, so the posterior predictive checks could be untrustworthy.
Furthermore, thinking about this situation with CV, I think leave-one-out is not what I want with matched pairs, but rather I want ‘leave-pair-out’ CV. Thus I decided to do leave-4 pairs-out K-fold CV, which should be a good estimate. Even leaving four pairs out takes forever to run, as K=44 here. But I run it and obtain these results:

Basically leave-4pairs-out CV is similar for both models, although p_kfold is much higher for the Poisson model, which is I believe due to the varying intercept for pairs acting in place of the dispersion parameter in the NB model. In fact, the estimate for the sd for the varying intercept for pairs in the Poisson model is higher than in the NB model.
As mentioned the results are similar (though certainly not identical) for the two models… I am leaning towards using the results from the NB model, simply based on past experience and the fact the model ran much better and the posterior predictive checks aren’t horrible. I would love for @avehtari or someone else to weigh in on these results. Is there a best model here? If I had to go with one model, which results for inference on the treatment effect should I use?
I was impressed with the difference in LOO and leave-pair-out K-fold CV…I guess not so surprising as they answer different questions, but I will admit being a little surprised.