Hi, I’m looking for suggestions on how to tackle low eff/low E-BFMI/Rhat issues in a multilevel model in an organized manner. I describe below my data and objective and what I have tried so far. Shall I try different prior distributions? A mixture of centered and non-centered parameterization? Is my model or code even wrong to start with? Is it due to the data?
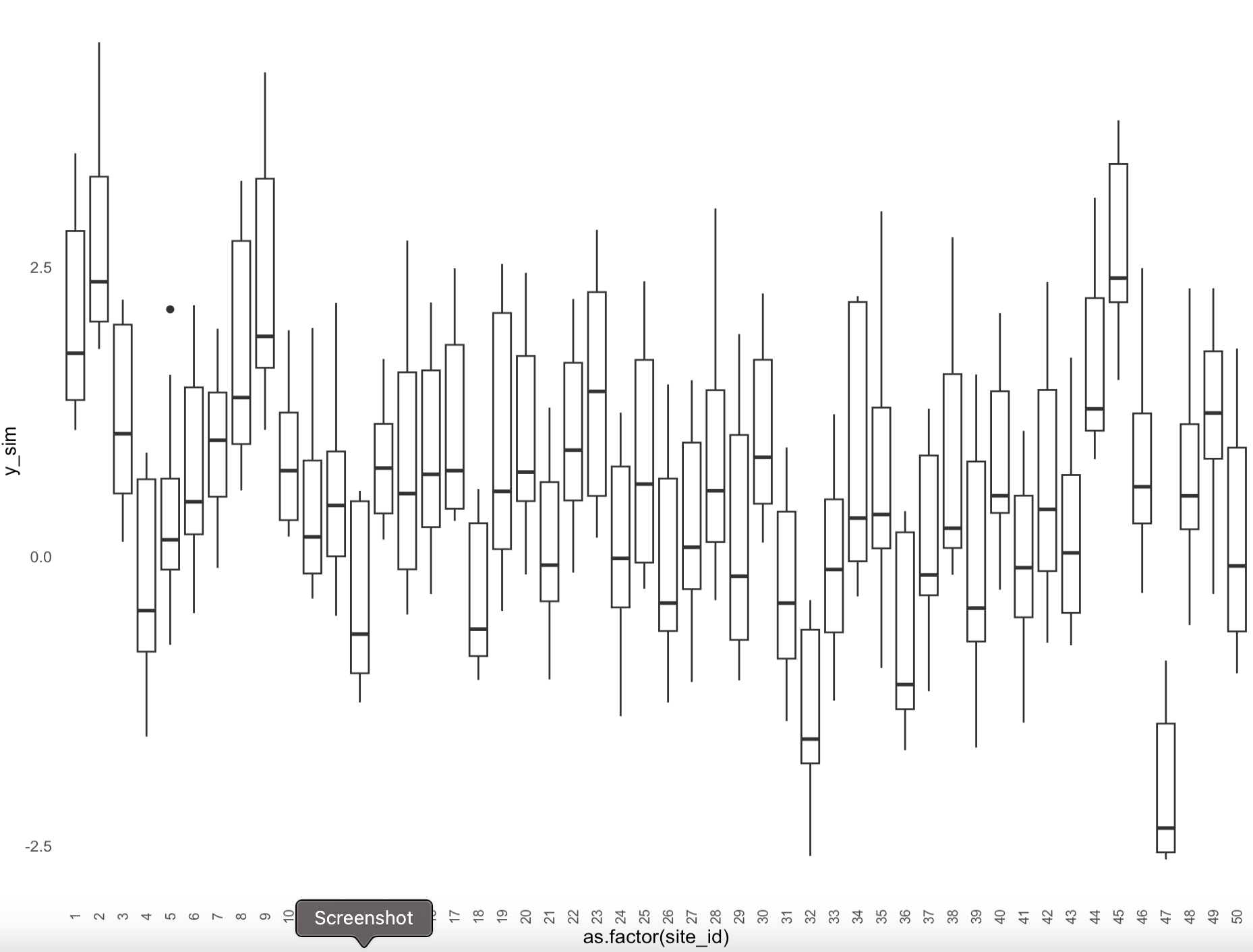
Data: ecological dataset with multiple levels related to the controlling variables of interest (“site” and “species”) and the sampling design (“plot”). On each of the 50 sites, there were 4 plots, on each plot we measured 3 species. The measurement is on a real scale (could be anything from -15 to +15), I standardized it for modeling purposes (center-scaled).
Objective: to check the effects of site and species. I expect “plot” to have no effect, it’s replication at the site scale.
Tried: the following multilevel model with centered parameterization, where \bar{\alpha} is the grand mean, \alpha the deviation from the grand mean due to site, and \beta the deviation due to species. Then \alpha + \beta would provide the expected distribution for the measured variable for a given species at a given site.
data {
int<lower=0> N; // number of observations
int<lower=0> N_site; // number of sites
int<lower=0> N_species; // number of species
int<lower=0> N_plot; // number of plots
array[N] int<lower=1> site; // vector of site idx
array[N] int<lower=1> species; // vector of species idx
array[N] int<lower=1> plot; // vector of plot idx
vector[N] y_obs; // vector of observed d15N values
}
parameters {
vector[N_site] alpha;
real alpha_bar;
real<lower=0> sigma_alpha;
vector[N_species] beta;
real<lower=0> sigma_beta;
vector[N_plot] gamma;
real<lower=0> sigma_gamma;
real<lower=0> sigma;
}
model {
// adaptive priors
target += normal_lpdf(alpha | alpha_bar, sigma_alpha);
target += normal_lpdf(beta | 0, sigma_beta);
target += normal_lpdf(gamma | 0, sigma_gamma);
target += exponential_lpdf(sigma | 1);
// hyper-priors
target += normal_lpdf(alpha_bar | 0, 10);
target += exponential_lpdf(sigma_alpha | 1);
target += exponential_lpdf(sigma_beta | 0.1);
target += exponential_lpdf(sigma_gamma | 0.5);
// likelihood
target += normal_lpdf(y_obs | alpha[site] + beta[species] + gamma[plot], sigma);
}
generated quantities {
// posterior predictive distribution for replications y_rep of the original data set y given model parameters
array[N] real y_obs_rep = normal_rng(alpha[site] + beta[species] + gamma[plot], sigma);
// compute log-lik
vector[N] log_lik;
for (i in 1:N) {
log_lik[i] = normal_lpdf(y_obs[i] | alpha[site[i]] + beta[species[i]] + gamma[plot[i]], sigma);
}
}
This centered parameterization leads to low E-BFMI, pretty bad “mcmc nuts energy” plots, highly correlated parameter estimates, and I can see some “departures” (not sure how to call that) in the trace and rank plots… The model reproduce the data pretty well though. I played a bit with the priors and they affect the spread of the posteriors, reasonably informative priors from expect knowledge help reducing issues but do not solve them all.
I then tried the non-centered parameterization below… but it creates divergences, persistent with increasing adapt_delta, so I assume it’s even worse.
data {
int<lower=0> N; // number of observations
int<lower=0> N_site; // number of sites
int<lower=0> N_species; // number of species
int<lower=0> N_plot; // number of plots
array[N] int<lower=1> site; // vector of site idx
array[N] int<lower=1> species; // vector of species idx
array[N] int<lower=1> plot; // vector of plot idx
vector[N] y_obs; // vector of observed d15N values
}
parameters {
real alpha_bar;
vector[N_site] a;
vector[N_species] b;
vector[N_plot] g;
real<lower=0> sigma_alpha;
real<lower=0> sigma_beta;
real<lower=0> sigma_gamma;
real<lower=0> sigma;
}
model {
// priors
target += normal_lpdf(alpha_bar | 0, 1);
target += normal_lpdf(a | alpha_bar, sigma_alpha);
target += normal_lpdf(b | 0, 1);
target += normal_lpdf(g | 0, 1);
target += exponential_lpdf(sigma_alpha | 1);
target += exponential_lpdf(sigma_beta | 1);
target += exponential_lpdf(sigma_gamma | 1);
target += exponential_lpdf(sigma | 1);
// likelihood
target += normal_lpdf(y_obs | alpha_bar + a[site] * sigma_alpha + b[species] * sigma_beta + g[plot] * sigma_gamma, sigma);
}
generated quantities {
// recover "decentered" parameters
vector[N_site] alpha = alpha_bar + a * sigma_alpha;
vector[N_species] beta = b * sigma_beta;
vector[N_plot] gamma = g * sigma_gamma;
// posterior predictive distribution for replications y_rep of the original data set y given model parameters
array[N] real y_obs_rep = normal_rng(alpha_bar + a[site] * sigma_alpha + b[species] * sigma_beta + g[plot] * sigma_gamma, sigma);
// compute log-lik
vector[N] log_lik;
for (i in 1:N) {
log_lik[i] = normal_lpdf(y_obs[i] | alpha[site[i]] + beta[species[i]] + gamma[plot[i]], sigma);
}
}