Dear all.
I’m trying to fit a quite complex semi-Bayesian human learning computational model to participant’s data (or simulated data).
The model itself is quite complex, and I use a user-defined function to code it, and this part works quite well. The simplest model has two free parameters - GD and LD. These parameters are generally limited from -1 to 1. When coding a simple STAN model for a single participant, I use Phi_approx with an additional transformation to reach these bounds, and it usually works quite well (good mixing). This the model I used for single participants that works:
parameters{
real GD0;
real LD0;
}
transformed parameters{
real GD;
real LD;
GD=2*(Phi_approx(GD0)-0.5);
LD=2*(Phi_approx(LD0)-0.5);
// the 2 and -0.5 are used to create parameters bounded from -1 to 1 instead of 0 to 1
}
model{
vector [nTrials] pRight1;
lamdaDepletion0~normal(0,0.3);
gammaDepletion0~normal(0,0.3);
// I used SD of 0.3 instead of 1 because I want to create a distribution with more mass on values around
zero, and low mass on values close to 1 and -1.
pRight1=Bmodel(dataValues,GD,LD);
//Bmodel is the model itself, and the other values are just data. It produces a single value - the probability of a right arrow response by a participant, which is then used for the bernoulli likelihood below.
Response~bernoulli(pRight1);
}
So this model works pretty well. However, when I try to implement a hierarchical model for all participants using non-central parameterization as follows I always get warnings regarding max_treedepth exceeded, and poor mixing (see below). This is how I am trying to implement the hierarchical model:
parameters{
real GD0[nSubj];
real LD0[nSubj];
real GD0_MU;
real LD0_MU;
real <lower=0> GD0_SD;
real <lower=0> LD0_SD;
}
transformed parameters{
vector [nSubj] GD;
vector [nSubj] LD;
GD=2*(Phi_approx(GD0_SD*to_vector(GD0)+GD0_MU)-0.5);
LD=2*(Phi_approx(LD0_SD*to_vector(LD0)+LD0_MU)-0.5);
}
model{
vector [nTrials] pRight1[nSubj];
LD0_MU~normal(0,0.3);
GD0_MU~normal(0,0.3);
LD0_SD~normal(0,0.1)T[0,];
GD0_SD~normal(0,0.1)T[0,];
LD0~normal(0,1);
GD0~normal(0,1);
for (s in 1:nSubj){
pRight1[s]=Bmodel(dataValues,GD[s],LD[s]);
Response[s]~bernoulli(pRight1[s]);
}
}
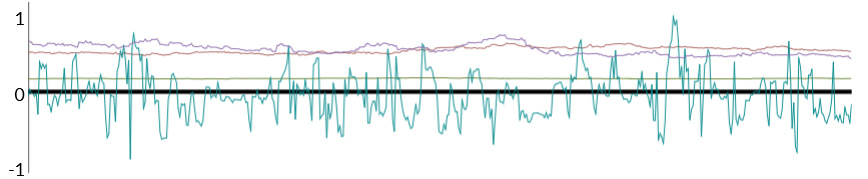
Here is an example of a typical result for the latter model (Clearly only Chain 3 works. The others just stay stuck, and do not mix). I’ve tried decreasing adapt_delta to 0.6, and increase max_treedepth - but that didn’t do the job. I even tried adding jitter to the step size - still did not work.
I appologize for not inserting the full model but it is very long, and I don’t think anyone would like to get into it just to answer my question. Nonetheless any help would be appreciated.

Thanks,
Isaac