Hi everyone,
I’m modelling the effects of protective characteristics on a deadly disease. I’m investigating the questions of whether characteristic 1 and characteristic 2 lower the risk of developing the disease altogether, whether they delay the disease’s onset and whether they affect number of months survived after diagnosis. In approximately 5-10% of cases, this disease has a genetic cause.
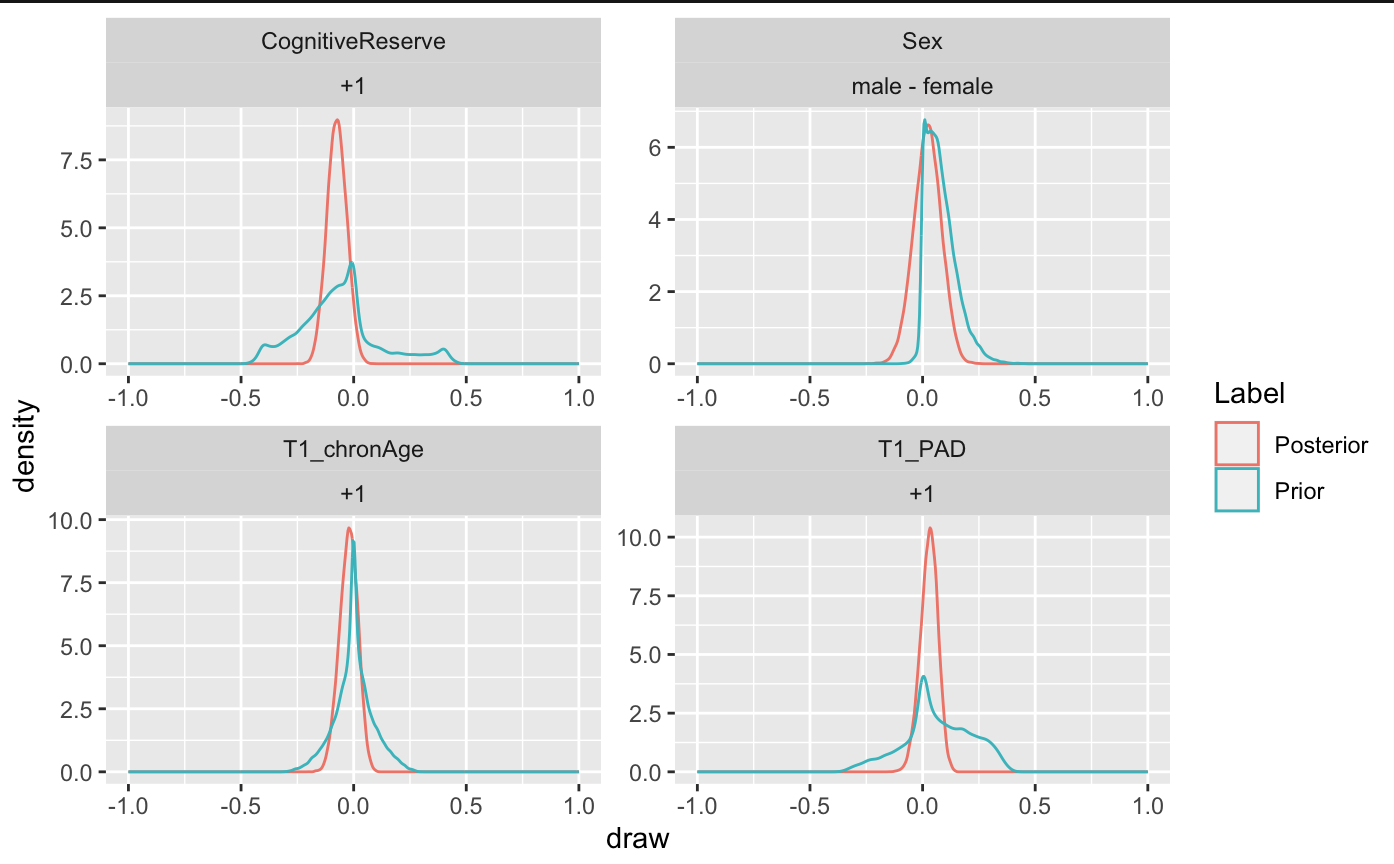
My questions are these: if I expect a characteristic to have an inverse relationship with the outcome and I want the majority of the prior to be negative, is normal(-1,1) or cauchy(-1,1) the right way to go to model my expectation of a negative effect? I ran my models this way, thinking it was correct, but when I produced figures comparing my priors and posteriors, the priors are still centered on 0.
We are using HC as the refcat, so we’re placing priors on the ALS patients. Here are my thoughts for this model:
-
CR: based on my previous work on CR’s effect, and what we know from Alzheimer’s disease, I expect
that higher CR is associated with a lower risk of developing ALS, consequently, I would set a slightly
negative prior (-1), with a standard deviation (SD) of 1; -
PAD: based on our previous work (Hermann et al., 2022), I would expect a higher PAD to be associated with a higher risk of developing ALS, and I will integrate this expectation into the model in the form of a positive prior with a large SD (centred at 1, SD of 2);
-
Age: I do not expect a major effect of age in this sample, so I center the prior on zero, with an SD of
1;
- Sex: We know that men have a higher risk of developing ALS, so I will set a prior reflecting this.
This is the code for the logistic regression of developing the disease, including the code to produce the graphs:
Model1_priors <- brm(data=df,
family = bernoulli("logit"),
ALSvsHC~1+T1_PAD+CognitiveReserve+T1_chronAge+Sex,
prior=c(set_prior("cauchy(-1,1)", class="b", coef="CognitiveReserve"),
set_prior("normal(1,2)", class="b", coef="T1_PAD"),
set_prior("normal(0,1)", class="b", coef="T1_chronAge"),
set_prior("normal(1,0.5)", class="b", coef="Sexmale")),
sample_prior = "only", #sampling the prior to compare prior to posterior distribution
iter=10000,
chains=4,
save_pars = save_pars(all = TRUE)) #if we wish to calculate the BF, we must save all parameters
Model1_posteriors <- brm(data=df,
family = bernoulli("logit"),
ALSvsHC~1+T1_PAD+CognitiveReserve+T1_chronAge+Sex,
prior=c(set_prior("normal(-1,1)", class="b", coef="CognitiveReserve"),
set_prior("normal(1,2)", class="b", coef="T1_PAD"),
set_prior("normal(0,1)", class="b", coef="T1_chronAge"),
3
set_prior("normal(1,0.5)", class="b", coef="Sexmale")),
iter=10000,
chains=4,
save_pars = save_pars(all = TRUE))
cmp <- list(
"Prior" = avg_comparisons(Model1_priors),
"Posterior" = avg_comparisons(Model1_posteriors))
draws <- lapply(names(cmp), \(x) transform(posteriordraws(cmp[[x]]), Label = x))
draws <- do.call("rbind", draws)
ggplot(draws, aes(x = draw, color = Label)) +
xlim(c(-1, 1)) +
geom_density() +
facet_wrap(~term + contrast, scales = "free")
Am I thinking about this correctly? Should I make any changes? And since genetics affect the probability of the disease, should I add a variable reflecting the information of “was tested, and positive for gene 1”, “was tested, and positive for gene 2”, “was tested and negative” and “was not tested”? If so, how could I best incorporate this information in my models?
Thank you in advance everyone!