Hi all,
I am in the process of building an occupancy model that uses data from amplicon sequencing (number of DNA sequences) as its detection model (see Conditional posterior predictions for three-level occupancy model for more details).
To explain the modelling problem, I will first explain briefly the data generating process, and the problem I am encountering. DNA is individually extracted from a set of samples. A marker gene (in my current case, 16S rRNA) is selected for amplification using PCR. The key detail here is that the PCR primers are tagged with a unique forward and reverse index sequence - there are 24 unique forward indices, and 24 unique reverse indexes, for 576 total combinations. Each DNA extract is assigned a unique combination of forward and reverse indices, which allows it to be re-identified later from the sequencing data. Once the samples have been amplified, they are combined together into a single pool of DNA for library preparation and then sequencing. The library is then sequenced, and the resulting sequences are processed through a pipeline that outputs a taxon by sample count matrix.
If this tagging process was perfect, there would be little problem. However, it turns out that during the library preparation, there is a small possibility that these tags to get ‘mixed up’ (called tag jumping or tag switching), which occurs when two molecules that are highly similar or identical, but with different tags, bind together:
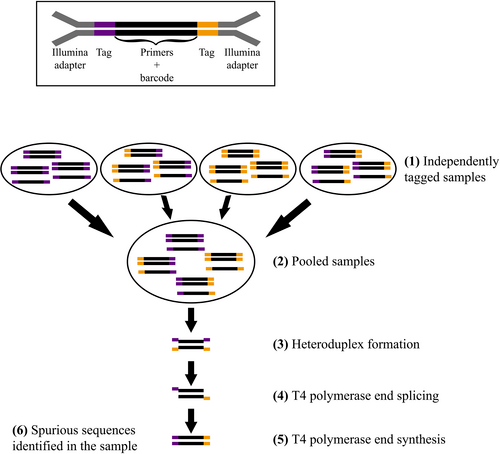
source: https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13745
This means that there is some level of unknown cross-contamination of samples that occurs during this step, and we can no longer assume that there are no false positives when occupancy modelling.
Luckily, we have data that might allow us to estimate these rates. Not every combination of tags is used, and so this process of tag jumping also recruits sequences into tag combinations that were not used during the PCR stage. In our datasets, we have used approximately 380 tag combinations of the 576, giving roughly 200 unused combinations for which we have data.
There are common ad-hoc methods for dealing with this problem, which typically involve estimating some threshold sequence count and subtracting it from the count matrix (with a floor of zero) - however, this does not seem particularly Bayesian as there will be uncertainty in the number of contaminant seqeunces in each sample.
I have been thinking about how to model this, to account for these false positive detections. Typically when modelling these kind of sequencing counts, a Poisson or Negative binomial is used, with some suitable offset (e.g. total read counts, or geometric means) to account for variation in overall sequencing depth. Here, we can then think of these counts as the sum of two sets of counts - an unknown number of true reads, and an unknown (smaller) number of contaminant reads.
Thinking in the Poisson case for a moment, I think this would allow estimation (for taxon j in sample i) as follows:
Where q is the (probably constant) rate of sequence contamination, and p is the true rate of seqence recruitment. This would probably require consideration of some kind of offset for both rates to account for varying sequencing depths.
And extending this to occupancy as follows, for used combinations, with occupancy probability \psi:
The problem is that in reality, the resulting counts are usually overdispersed, and a Poisson fits poorly if at all, so a Negative Binomial with some mean \mu and dispersion \phi is chosen to model these counts. I believe it should be possible to do the same sum of rates as above with a Negative Binomial, but only if the dispersion parameters are equal. However, I don’t think it would be reasonable to expect this is the case - it might even be that the contamination is more closely Poisson distributed, though I haven’t done any empirical poking at this yet.
Can anyone suggest a count distribution, or a method for combining count distributions, that might be appropriate here? I have been thinking about this for a little while, and I am concerned that this could easily be intractable if it requires marginalising over some unknown number of counts (the biggest number of sequences in an unused combination is just over 600).



